中央研究院 生物化學研究所
中央研究院 生物化學研究所
中央研究院 生物化學研究所
中央研究院 生物化學研究所
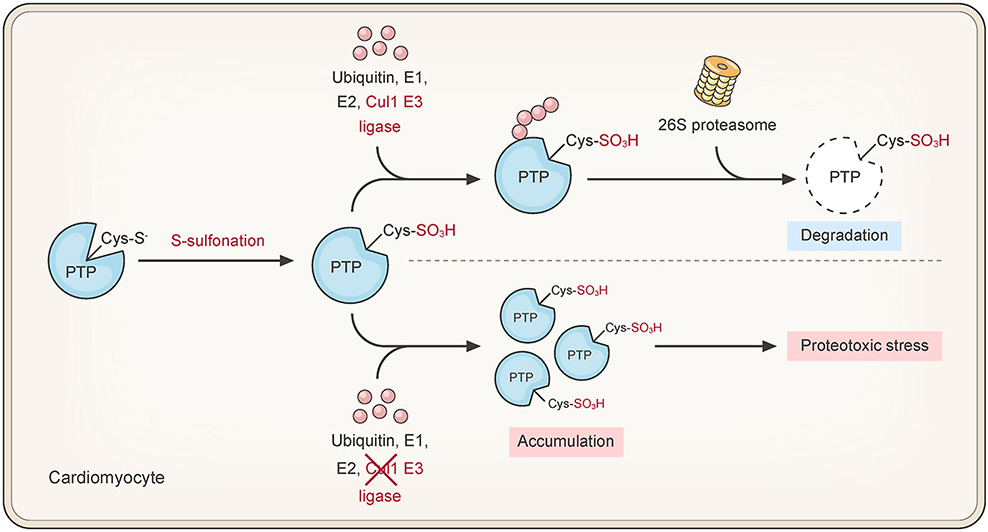
Reactive oxygen species (ROS), released as byproducts of mitochondrial metabolism or as products of NADPH oxidases and other processes, can directly oxidize the active-site cysteine (Cys) residue of protein tyrosine phosphatases (PTPs) in a mammalian cell. Robust degradation of irreversibly oxidized PTPs is essential for preventing accumulation of these permanently inactive enzymes. However, the mechanism underlying the degradation of these proteins was unknown. In this study, we found that the active-site Cys215 of endogenous PTP1B is sulfonated in H9c2 cardiomyocytes under physiological conditions. The sulfonation of Cys215 led PTP1B to exhibit a conformational change, and drive the subsequent ubiquitination and degradation of this protein. We then discovered that Cullin1, an E3 ligase, interacts with the Cys215-sulfonated PTP1B. The functional impairment of Cullin1 prevented PTP1B from oxidation-dependent ubiquitination and degradation in H9c2 cells. Moreover, delivery of the terminally oxidized PTP1B resulted in proteotoxicity-caused injury in the affected cells. In conclusion, we elucidate how sulfonation of the active-site Cys215 can direct turnover of endogenous PTP1B through the engagement of ubiquitin-proteasome system. These data highlight a novel mechanism that maintains PTP homeostasis in cardiomyocytes with constitutive ROS production.