中央研究院 生物化學研究所
中央研究院 生物化學研究所
中央研究院 生物化學研究所
中央研究院 生物化學研究所
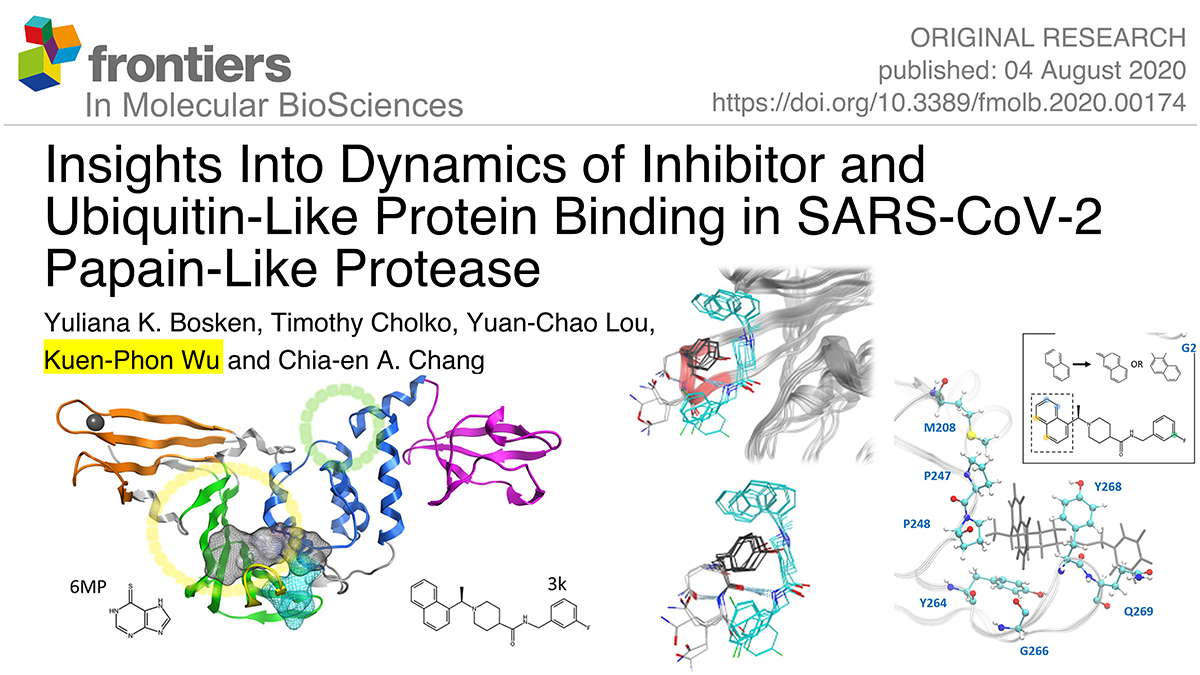
Covid-19 is caused by a novel form of coronavirus for which there are currently no vaccines or anti-viral drugs. This virus, termed SARS-CoV-2 (CoV2), contains Papain-like protease (PLpro) involved in viral replication and immune response evasion. Drugs targeting this protease therefore have great potential for inhibiting the virus, and have proven successful in older coronaviruses. Here, we introduce two effective inhibitors of SARS-CoV-1 (CoV1) and MERS-CoV to assess their potential for inhibiting CoV2 PLpro. We ran 1 μs molecular dynamics (MD) simulations of CoV2, CoV1, and MERS-CoV ligand-free PLpro to characterize the dynamics of CoV2 PLpro, and made comparisons between the three to elucidate important similarities and differences relevant to drug design and ubiquitin-like protein binding for deubiquitinating and deISGylating activity of CoV2. Next, we simulated the inhibitors bound to CoV1 and CoV2 PLpro in various poses and at different known binding sites to analyze their binding modes. We found that the naphthalene-based ligand shows strong potential as an inhibitor of CoV2 PLpro by binding at the putative naphthalene inhibitor binding site in both computational predictions and experimental assays. Our modeling work suggested strategies to improve naphthalene-based compounds, and our results from molecular docking showed that the newly designed compounds exhibited improved binding affinity. The other ligand, chemotherapy drug 6-mercaptopurine (6MP), showed little to no stable intermolecular interaction with PLpro and quickly dissociated or remained highly mobile. We demonstrate multiple ways to improve the binding affinity of the naphthalene-based inhibitor scaffold by engaging new residues in the unused space of the binding site. Analysis of CoV2 PLpro also brings insights into recognition of ubiquitin-like proteins that may alter innate immune response.