中央研究院 生物化學研究所
中央研究院 生物化學研究所
中央研究院 生物化學研究所
中央研究院 生物化學研究所
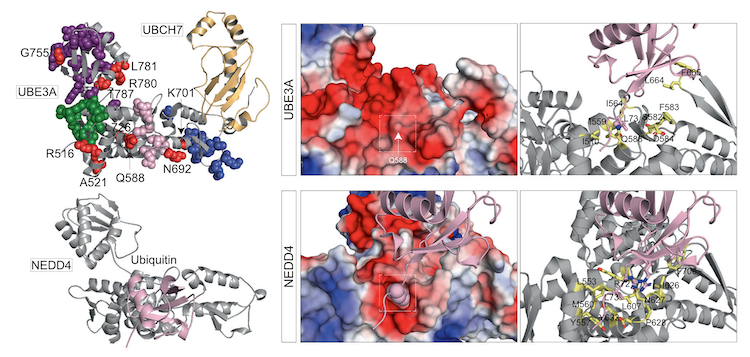
The mechanisms that underlie the extensive phenotypic diversity in genetic disorders are poorly understood. Here, we develop a large-scale assay to characterize the functional valence (gain or loss-of-function) of missense variants identified in UBE3A, the gene whose loss-of-function causes the neurodevelopmental disorder Angelman syndrome. We identify numerous gain-of-function variants including a hyperactivating Q588E mutation that strikingly increases UBE3A activity above wild-type UBE3A levels. Mice carrying the Q588E mutation exhibit aberrant early-life motor and communication deficits, and individuals possessing hyperactivating UBE3A variants exhibit affected phenotypes that are distinguishable from Angelman syndrome. Additional structure-function analysis reveals that Q588 forms a regulatory site in UBE3A that is conserved among HECT domain ubiquitin ligases and perturbed in various neurodevelopmental disorders. Together, our study indicates that excessive UBE3A activity increases the risk for neurodevelopmental pathology and suggests that functional variant analysis can help delineate mechanistic subtypes in monogenic disorders.