中央研究院 生物化學研究所
中央研究院 生物化學研究所
中央研究院 生物化學研究所
中央研究院 生物化學研究所
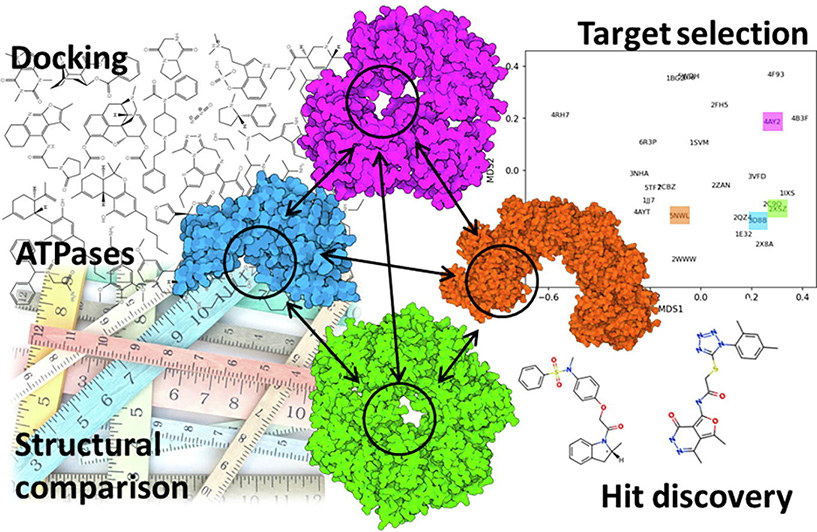
The ATP binding sites of many enzymes are structurally related, which complicates their development as therapeutic targets. In this work, we explore a diverse set of ATPases and compare their ATP binding pockets using different strategies, including direct and indirect structural methods, in search of pockets attractive for drug discovery. We pursue different direct and indirect structural strategies, as well as ligandability assessments to help guide target selection. The analyses indicate human RAD51, an enzyme crucial in homologous recombination, as a promising, tractable target. Inhibition of RAD51 has shown promise in the treatment of certain cancers but more potent inhibitors are needed. Thus, we design compounds computationally against the ATP binding pocket of RAD51 with consideration of multiple criteria, including predicted specificity, drug-likeness, and toxicity. The molecules designed are evaluated experimentally using molecular and cell-based assays. Our results provide two novel hit compounds against RAD51 and illustrate a computational pipeline to design new inhibitors against ATPases.