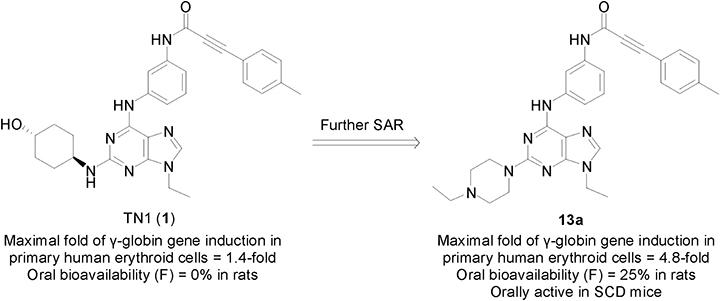
Reactivation of fetal hemoglobin (HbF) expression by therapeutic agents has been suggested as an alternative treatment to modulate anemia and the related symptoms of severe β-thalassemia and sickle cell disease (SCD). Hydroxyurea (HU) is the first US FDA-approved HbF inducer for treating SCD. However, approximately 25% of the patients with SCD do not respond to HU. A previous study identified TN1 (1) as a small-molecule HbF inducer. However, this study found that the poor potency and oral bioavailability of compound 1 limits the development of this inducer for clinical use. To develop drug-like compounds, further structure-activity relationship studies on the purine-based structure of 1 were conducted. Herein, we report our discovery of a more potent inducer, compound 13a, that can efficiently induce γ-globin gene expression at non-cytotoxic concentrations. The molecular mechanism of 13a, for the regulation HbF expression, was also investigated. In addition, we demonstrated that oral administration of 13a can ameliorate anemia and the related symptoms in SCD mice. The results of this study suggest that 13a can be further developed as a novel agent for treating hemoglobinopathies, such as β-thalassemia and SCD.

 Institute of Biological Chemistry, Academia Sinica
Institute of Biological Chemistry, Academia Sinica